Persistent Embryo Method and Landau-Ginzburg-Devonshire Model for Domain Nucleation¶
©️ Copyright 2025 @ Liu Theory Lab
Author:
Jiyuan Yang
Date:2025-10-20
Lisence:This document is licensed under Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0) license.
In this tutorial I will show how to use revised Persistent Embryo Method (rPEM) to determine the critical nucleus during ferroelectric switching, and explain the Landau-Ginzburg-Devonshire (LGD) Model based on the results of molecular dynamics (MD) simulations.
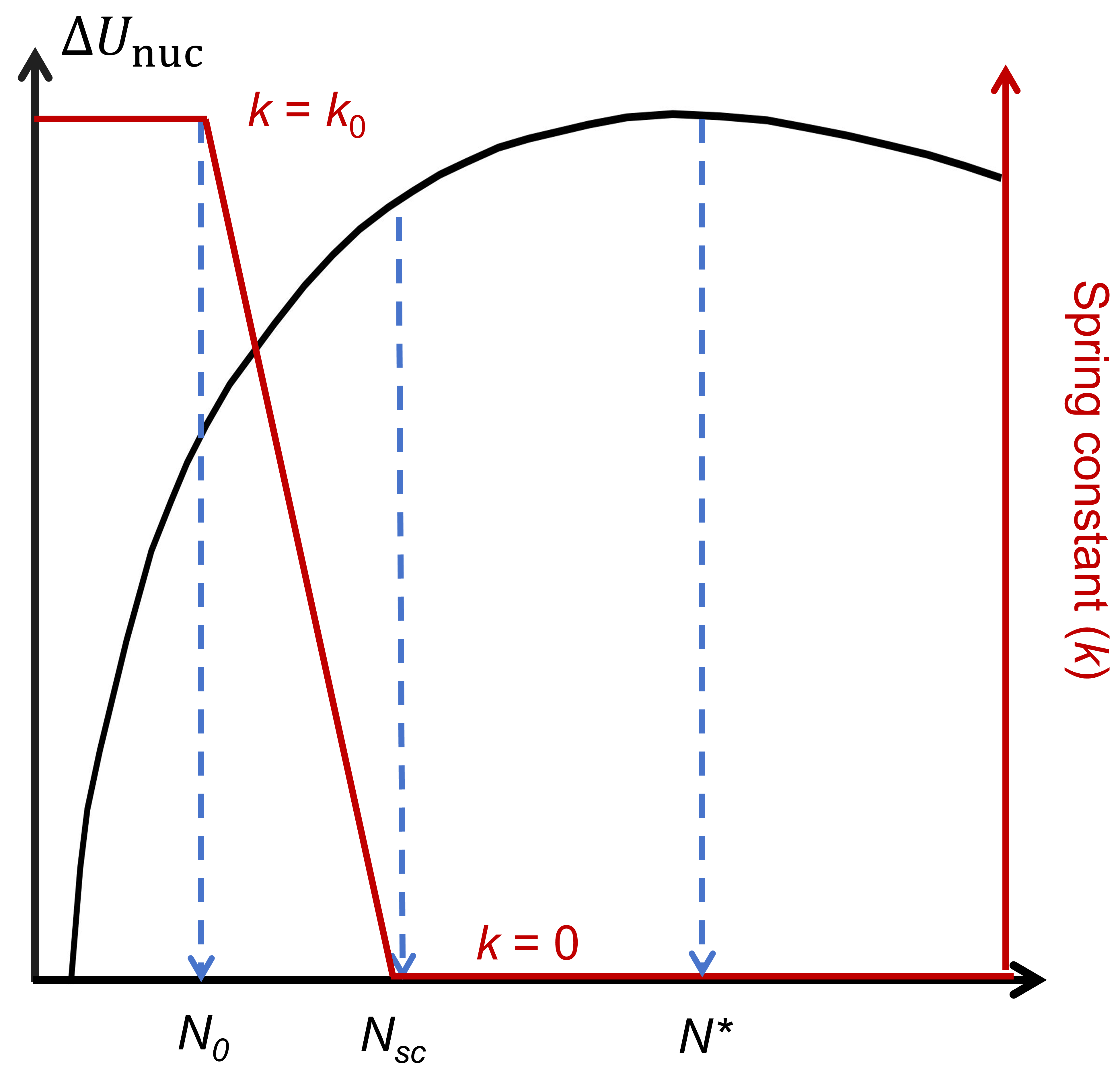
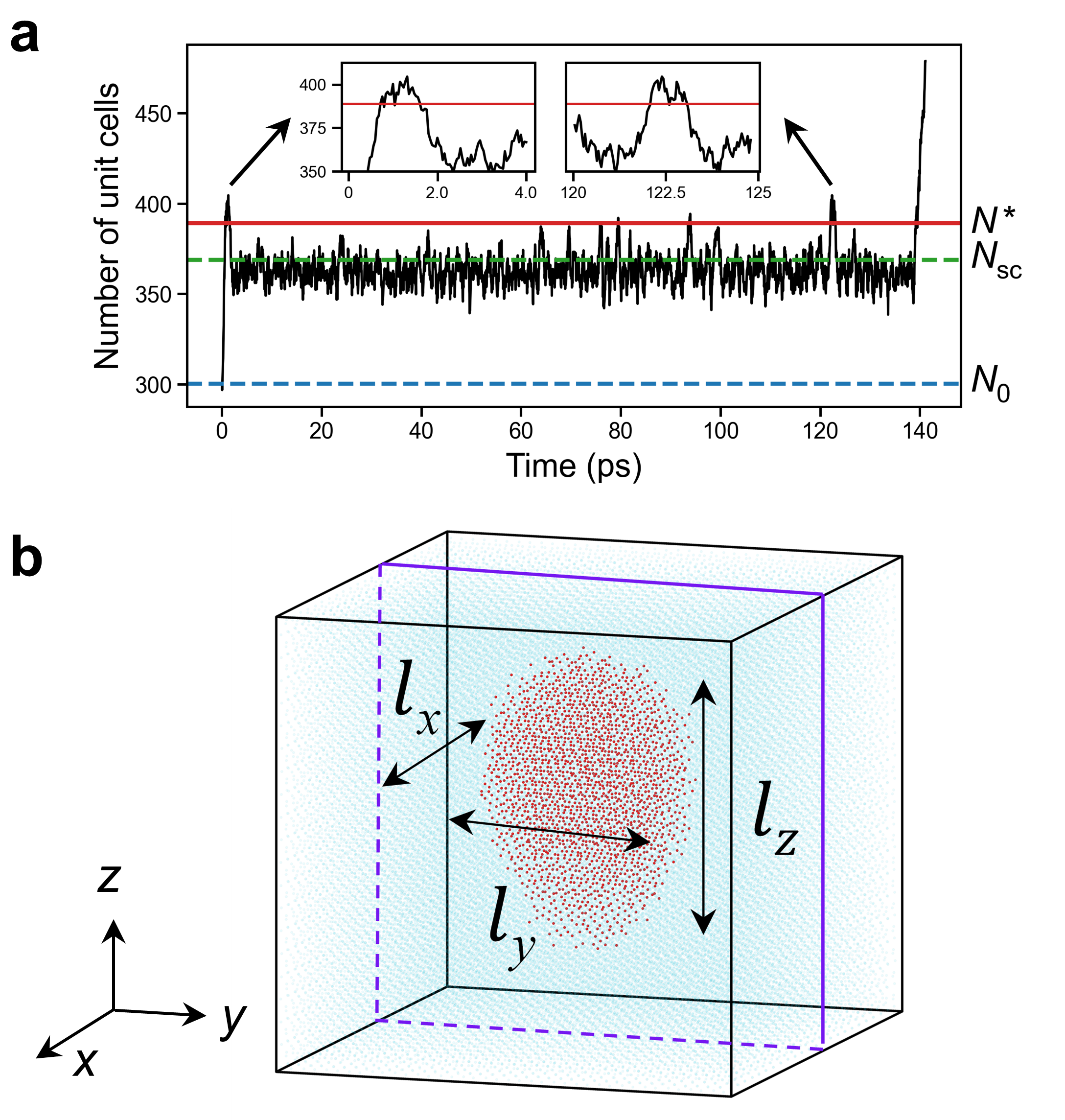
Traditional molecular dynamics simulations have limitations in capturing the nucleation of reversed domains under weak electric fields, as such events are extremely rare on accessible timescales. To address this issue, we employ PEM, originally developed for simulating three-dimensional (3D) crystal nucleation in liquids. Our improved version, termed the rPEM method, is specifically designed for simulating electric-field-driven nucleation in ferroelectrics. In the rPEM method, a 3D embryo consisting of \(N_0\) reversed oxygen atoms (or unit cells) is first embedded within a single-domain structure.
To prevent the system from reverting to its original state, an adjustable harmonic potential is applied to each oxygen atom in the embryo. This strategy effectively suppresses long-term nucleation fluctuations present in smaller nucleus. Once the nucleus size \(N\) exceeds the subcritical threshold \(N_{sc}\), the potential is gradually weakened and eventually removed. The elastic constant of the harmonic potential follows the formula \(k(N) = k_0\left[(N_{\rm sc} - N)/N \right]\) (when \(N < N_{\rm sc}\)), otherwise \(k(N) = 0\). By adaptively reducing the spring constant to zero before reaching the critical nucleus size, rPEM ensures that the system's dynamics at the critical point evolve naturally, i.e., without any bias. The observed plateau in the nucleus size evolution curve \(N(t)\) indicates the period during which the nucleus size oscillates around \(N^*\). These fluctuations allow us to determine the shape and size of the critical nucleus in an unbiased environment.
Take HfO\(_2\) as an example, here I show the rPEM code for 3D nucleation.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 | |
Then I will explain the key points in the code to make it easier to understand.
First you should install the plugin developed by Denan to enable displacement calculations in LAMMPS. The Github website for this plugin is: https://github.com/MoseyQAQ/liugroup-lammps-plugin.
Then use the following code to enable it in rPEM code:
The required LAMMPS plugin for displacement calculations has been pre-installed on the GPU computing cluster maintained by Liu's research group (administered by Lintao). Users can directly access this implementation using the following code without additional installation steps.
Then look at the MD settings: nloop determines when the rPEM algorithm terminates, kspr0 is the magnitude of the spring constant (driving force) that prevents back switching, time_window is the interval for adjusting kspr0. Nsc is the nucleus size when the driving force is canceled. Nbreak is the nucleus size used to determine complete reversal. The entire program stops when either Nbreak or nloop is reached. The rcut parameter determines the maximum interatomic distance for identifying atoms belonging to the same nucleus cluster. The initial reversed domain is defined within the spatial boundaries: X \(\in\) (Xmin, Xmax), Y \(\in\) (Ymin, Ymax), Z \(\in\) (Zmin, Zmax). The ave_disp_layer24 parameter specifies the characteristic displacement distance for oxygen atoms during reversed domain construction.
To start, first construct a reversed nucleus:
define the atoms not reversed:
Here we define the time-dependent size of nucleus on-the-fly, using the dynamic group:
Then we perform a quick energy minimization step to optimize the atomic configuration containing the initial nucleus structure.
After routinely applying the electric field, we have completed all preparations to initiate the rPEM cycle. First, save the state of the initial seed.
use following code to extract it from lmp.log file then plot the data:
LGD 3D nucleation model¶
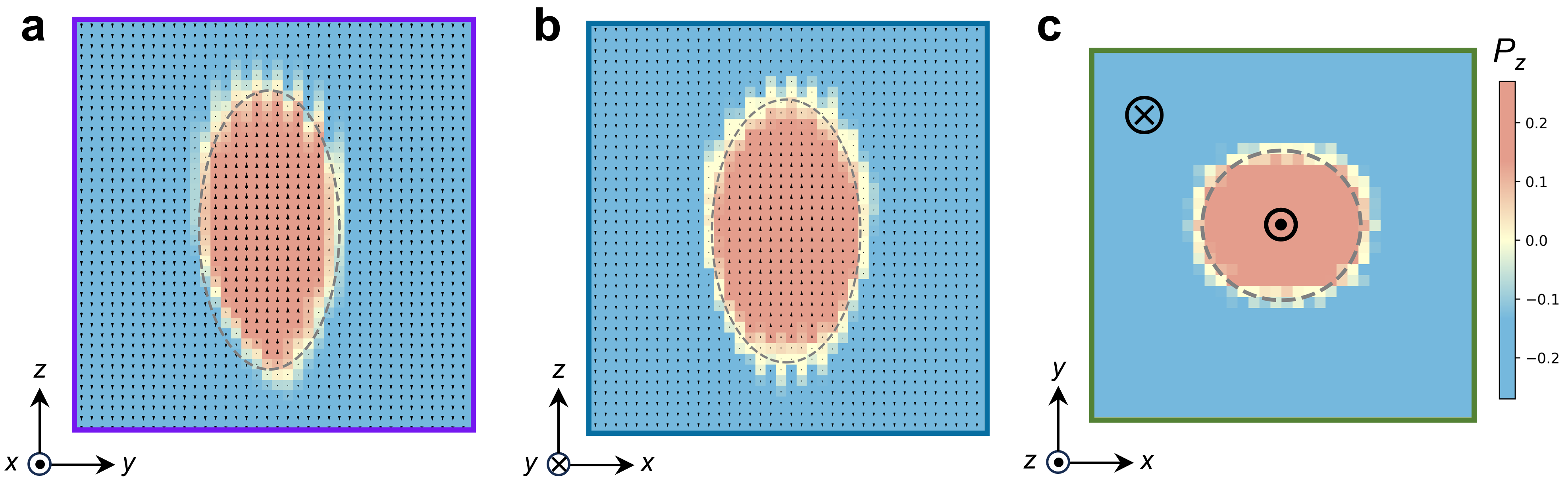
Based on the shape of nucleus from MD simulations, we can build a LGD model:
\(\Delta U_{\rm nuc} = -\frac{\pi}{3}l_x^2l_z\eta P_s\mathcal{E}+\int_{0}^{2\pi}\mathrm{d}\varphi\int_{0}^{\pi}\sigma_x\sqrt{\sin^2\theta+\left(\frac{\sigma_z}{\sigma_x}\right)^2\cos^2\theta}~(l_\theta/2)^2\sin\theta\mathrm{d}\theta\)
The details of this model can be found in PHYSICAL REVIEW X 15, 021042 (2025). Here I will use simple words to help readers understand the physical meaning of this model. Let us first look at the fully reversed (red) region. This region possesses the same energy as the initial blue region, meaning it incurs no additional energy penalty. Its sole effect is to reduce the total energy of the entire system by the following amount:
\(\Delta U_{\mathrm{V}} = - \mathcal{E} \eta \iiint_V\mathrm{d}V \left (P_z^{\rm nuc}(x,y,z)-P_z^{\rm SD}(x,y,z) \right)\)
Then look at the yellow region (the shell of nucleus). This region deviates from the system's energy optimum, thus incurring an energy penalty. The energy increase caused by the creation of these new surfaces can be expressed by the following equation:
\(\Delta U_{\mathrm{I}} =\iint_{S}\sigma_\theta\mathrm{dS}\) where \(\sigma_\theta\) is a spatial dependent surface energy along the shell.
So the energy of nucleus is the sum of these two energies:
\(\Delta U_{\mathrm{nuc}}=\Delta U_{\mathrm{V}}+\Delta U_{\mathrm{I}}\)
Note that this model is a zero-kelvin model, we need to extend it to finite temperature.
Use the method developed in Supplemental Material of PHYSICAL REVIEW X 15, 021042 (2025), we have:
\(\sigma_\theta = \frac{8P_s}{3}\sqrt{g_{\theta}K_\mathrm{loc}}\)
Therefore, the whole equation in fact is a function of \(P_s\). Note that we have the temperature dependent polar mode from Landau phase transition theory:
\(P_s(T)=\sqrt{\frac{T_c-T}{T_c}}P_s\)
So the free energy is conveniently calculated using a temperature dependent polar mode.
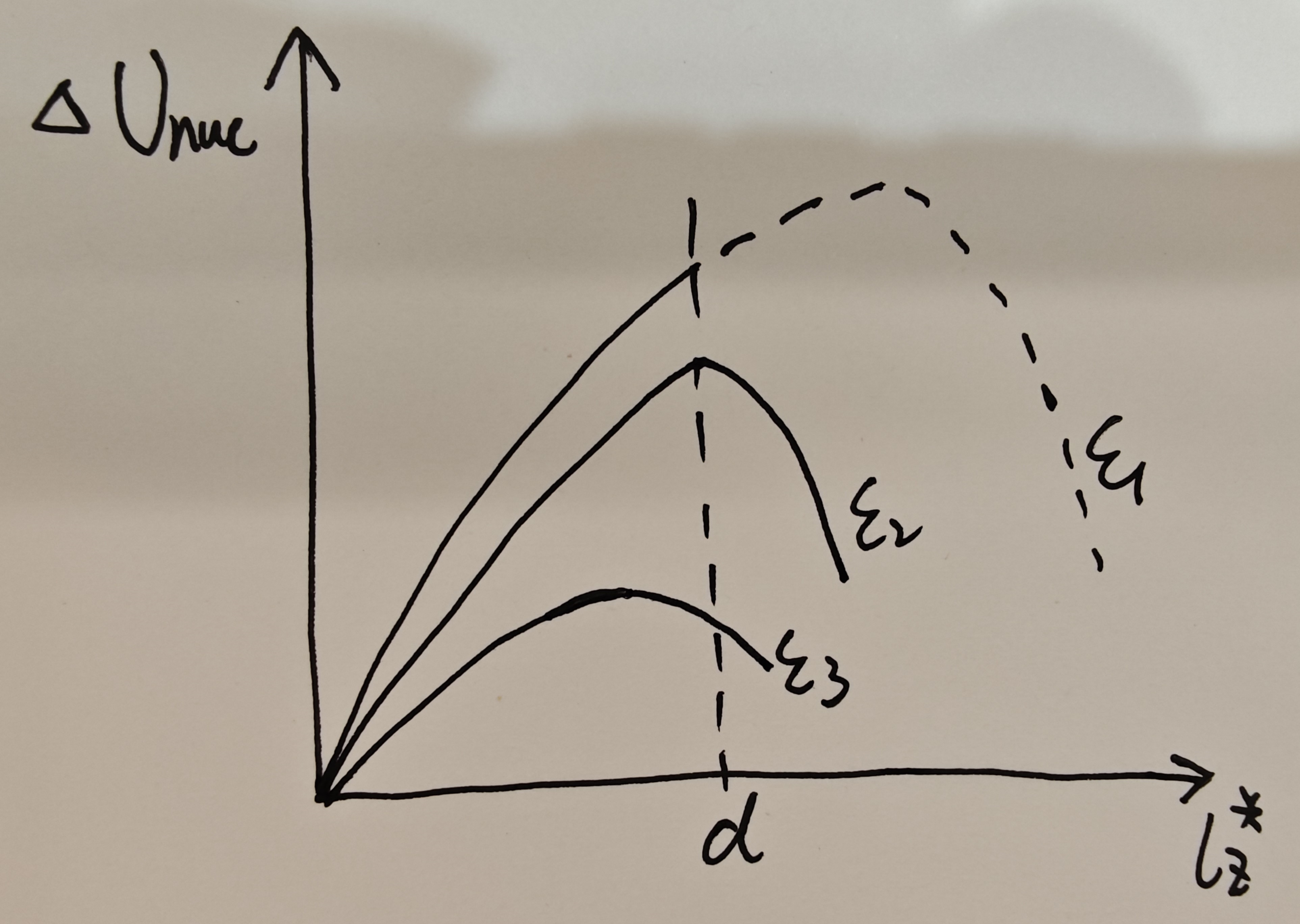
At a give electric field \(\mathcal{E}\) , our LGD model fast predicts the critical size \(𝑙_𝑧^∗(\mathcal{E})\) by calculating saddle point of \(\Delta U_{\mathrm{nuc}}(T)\).
This physical quantity measures the ease of nucleation. Consider a film with a thickness of d. When a low electric field \(\mathcal{E}_1\) is applied, the entire spatial extent of the film is insufficient to allow the system to reverse within the experimental timeframe. Only when the electric field is sufficiently large (e.g., \(\mathcal{E}_3\)) can nucleation and reversal occur within the thickness d. Thus, the coercive field is determined by the critical field when \(𝑙_𝑧^∗(\mathcal{E}_c)≈𝑑\).