AIRSS Workflow¶
©️ Copyright 2025 @ Liu Theory Lab
Author:
Ziyue Lin 📨
Date:2025-10-20
Lisence:This document is licensed under Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0) license.
I. Structure Search¶
1. Generate Files¶
Example: gencell 10 1 Rb 1 F 1
(Parameters like 10, 1, 1 can be changed later; first create the .cell file)
Parameters in order: - gencell: File generation command - 10: Preset volume - 1: Supercell multiplier - Rb, F: Element names composing the compound - 1, 1: Ratio of each element
This will generate two files: *.cell and *.param
2. Edit Files¶
(1) *.cell¶
Diagonal values are the cube root of volume
Volume, search at https://uspex-team.org/online_utilities/volume_estimation/
NUM is the number of each atom in the formula
Double ## represents comments; keeping only one # activates them. - First item: element types - Second item: total number of atoms - Third item: if activated, predicts structures containing pure elements Using the second item can delete the entire preceding %BLOCK LATTICE_CART parameter
(2) *.param¶
Cutoff energy: Drag the generated .cell file into VESTA, then export as .cif file and drag into Material Studio. Note: must be an English path. Select the three wavy lines icon Calculation-Electronic, options:
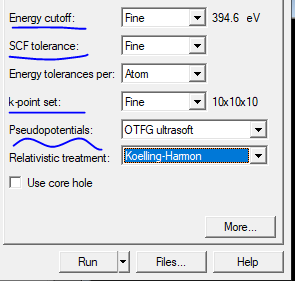
For Energy cutoff, SCF tolerance, k-point set, select Fine.
For Pseudopotentials, select On the Fly or OTFG ultrasoft.
Then check the value next to Energy cutoff; this value is the *.param cut_off_energy value.
When resources allow, actual calculations generally tend to be slightly higher than that number. Higher values correspond to higher accuracy, but computation speed will be slower.
You can also use MS for Encut testing: build→symmetry→find symmetry→click and then click find symmetry in new window→impose symmetry
3. airss.pbs¶
You can copy from others' working directories and modify (recommended), or create your own, but it varies slightly between servers. Example:
Supercomputing Center:
4. Results Analysis¶
Fixed cell:¶
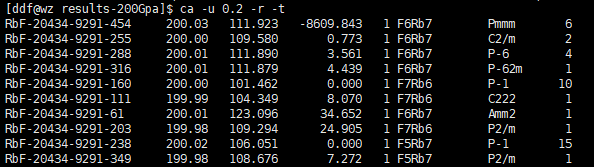
Finally, find the corresponding structure and export using Xftp or Winscp.
Note: Accuracy generally can be between 0.1 - 0.5. As for selecting how many structures for high-precision optimization, there's no unified number; depends on the enthalpy difference between them. Within 0.5eV should be considered.
- First column: Structure name label assigned by AIRSS software
- Second column: Pressure value (GPa)
- Third column: Volume per formula unit (fu)
- Fourth column: First row is enthalpy per formula unit (fu); subsequent rows are enthalpy values relative to the first row under different structures
- Fifth column: Total number of formula units (fu) in unit cell (number of fug in unit cell multiplied by number of fu in one fug)
- Sixth column: Chemical formula of formula unit (fu)
- Seventh column: Space group name
- Eighth column: Number of times this structure appears in all search results
If you think there are too many listed results, you can use the -u option, but note that -u must be used before -r.
Variable cell:¶
Command to list structures on convex hull plot:
Fixed cell:
Variable cell:
(The 0 at the end is accuracy; if you input 0.1, it lists points within 0.1 error)
Plot convex hull:
Additional Commands¶
After structure optimization:¶
- Copy res files found by airss structure search to another new folder called test
- Then input
ca -m -de 0.04 -r --deleteto delete res files with single atom energy difference greater than 0.04eV. The value 0.04 should vary based on actual situation - Copy the cell and param files from structure search to this new folder test, increase K-points in cell, add pressure, increase cutoff energy in param file
- Use command
run.pl -mpinp 8 -keepto continuously calculate all structures in folder test - Compare enthalpy values of these structures; the lowest enthalpy is the most stable structure
Other commonly used commands:¶
First create tmp folder
Only list the most stable stoichiometry and copy to tmp folder
Find the top ten stable structures of a certain stoichiometry and copy to tmp folder